
 Discussion
Discussion
Pompe disease symptoms
Last updated Nov. 7, 2024, by José Lopes, PhD
 Fact-checked by Patrícia Silva, PhD
Fact-checked by Patrícia Silva, PhD
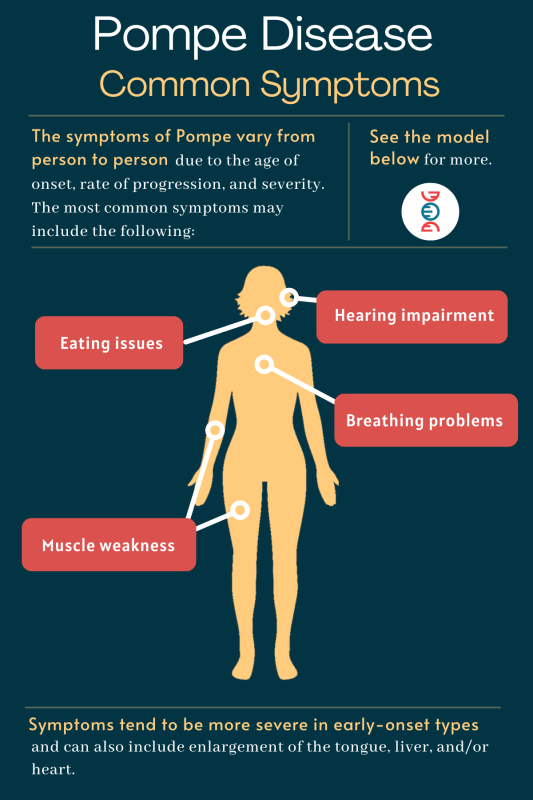
Pompe disease is a genetic disorder caused by mutations in the GAA gene, which results in progressive muscle weakness and other symptoms such as problems with eating, hearing, and breathing.
The symptoms of Pompe disease vary according to the age of onset, with the most severe seen in babies with the classic infantile-onset form of the disease. A faster rate of Pompe disease progression is associated with an earlier age of symptom onset.
Main symptoms of Pompe disease
There are a range of Pompe disease symptoms and the age of onset, rate of progression, disease severity, and disease type can all affect their severity.
The most common symptoms of Pompe include:
- progressive muscle weakness
- poor muscle tone
- muscle pain
- fatigue
- breathing problems
- respiratory infections
- trouble eating
- enlargement of the tongue, liver (hepatomegaly), and/or heart (cardiomegaly)
- scoliosis, where the spine twists and curves to the side, and hyperlordosis (an exaggerated inner curve of the lower spine)
- hearing impairment.
How symptoms are affected by Pompe disease type
Pompe disease is divided into three types, based on when symptoms manifest:
- classic infantile-onset
- non-classic infantile-onset
- late-onset.
In general, symptoms tend to be more severe in patients whose symptoms appear earlier.
Classic infantile-onset Pompe disease symptoms
Babies with classic infantile-onset Pompe will experience rapidly progressing symptoms from the first few months of life. These can include:
- muscle weakness and poor muscle tone, such that these children are sometimes referred to as floppy babies
- feeding difficulties and trouble growing and gaining weight at normal rates, a condition known as failure to thrive
- a large, protruding tongue (macroglossia)
- an enlarged liver
- hearing problems
- breathing issues
- resting the legs in a frog-like position, a characteristic sign of Pompe
- hypertrophic cardiomyopathy — when the heart muscle becomes thickened and cannot pump blood properly — which usually appears within the first weeks of life
- not being able to sit up, crawl, or stand.
If untreated, babies with this form of Pompe disease usually die within two years of birth. However, advances in treatment are improving outcomes.
Non-classic infantile-onset Pompe disease symptoms
Usually first appearing by age 1, non-classic infantile-onset Pompe disease symptoms can include:
- progressive muscle weakness and failing to hit motor milestones such as rolling over or sitting
- enlarged heart (cardiomegaly), but not heart disease (cardiomyopathy) in the first year of life, which distinguishes non-classic from classic infantile-onset disease
- serious breathing problems.
Without treatment, children with non-classic infantile Pompe disease often survive until early childhood, although treatment may improve outcomes.
Late-onset Pompe disease symptoms
Late-onset disease encompasses all forms of Pompe in which symptoms develop after the first year of life. The specific age at onset varies greatly from person to person; some people begin having symptoms in childhood, while others don’t experience symptoms until later in life. Generally, an earlier age at onset is associated with a faster rate of disease progression.
Late-onset Pompe disease symptoms can include:
- progressive weakness in proximal muscles, which are the muscles that are closer to the trunk of the body, including the shoulders, hips, upper arms, and upper legs; usually, the legs are more prominently affected than the arms
- muscle weakness in the face and throat, which can cause droopy eyelids (ptosis) and problems eating and swallowing, leading to gradual weight loss
- varying degrees of respiratory problems and frequent chest infections due to weakness of the muscles involved in breathing
- scoliosis and/or hyperlordosis
- profound fatigue
- chronic muscle pain and cramps
- hearing loss, an example of a Pompe disease neurological symptom
- headaches
- heart problems such as an abnormal heart rhythm (arrhythmia)
- enlarged heart
- digestive complaints.
Scroll horizontally to view all columns -->
| Disease type | Age at onset | Common symptoms |
|---|---|---|
| Classic infantile-onset | Within months of birth |
|
| Non-classic infantile-onset | By 1 year old |
|
| Late-onset | After age 1 |
|
Rare symptoms of Pompe disease
Besides the hallmark complications like progressive muscle weakness, low muscle tone, and breathing issues, people with Pompe disease may also experience rarer symptoms that affect multiple body parts, such as:
- rigid spine syndrome, characterized by contractures of spinal muscles associated with abnormal posture that limits neck and trunk movements
- hypothyroidism, when the thyroid gland doesn’t produce enough thyroid hormone
- gastrointestinal, or digestive issues, such as diarrhea and bowel incontinence
- blood vessel involvement, such as greater stiffness and dilation of arteries, as well as signs of blood vessel disease (vasculopathy) in the brain
- cognitive issues that affect the ability to plan ahead and meet goals, among other processes
- numbness or painful tingling or burning in the extremities, a disorder called small fiber neuropathy
- eye-related disorders, apart from ptosis, such as strabismus (eye misalignment) and ophthalmoplegia, or paralysis of eye muscles.
Early signs to look out for
Pompe disease is a multisystem disorder, meaning several organs may be affected besides muscle involvement, which is more well-known. In fact, the first symptoms of the disorder are as diverse as they are variable from patient to patient.
The factors that determine which early signs of Pompe disease a patient may present include their age at disease onset and their Pompe type. How fast the disease progresses is associated with how early in life symptoms appear and has an effect on Pompe disease life expectancy.
Infants
Infants who have symptoms very early in life, particularly those with the classic infantile-onset form, are the most severely affected among the Pompe disease patient population. In such cases, the earliest signs and symptoms usually present in the first months of life. Pompe disease symptoms in babies may include:
- rapidly progressive muscle weakness and hypotonia, or low muscle tone (floppiness)
- head lag, which refers to when the head seems to flop around or lags behind the trunk during a specific (pull-to-sit) maneuver
- delays in or not achieving milestones such as rolling over, sitting up, or standing
- breathing impairment
- feeding and swallowing problems
- poor weight gain and growth
- hypertrophic cardiomyopathy and enlarged heart
- larger than normal, protruding tongue.
Children and adults
The first signs and symptoms of Pompe disease in adults and older children may also affect several different body parts and functions. The first manifestations in these patients include:
- exercise intolerance and fatigue
- muscle pain, cramps, and muscle weakness, particularly in pelvic muscles
- gait disturbances, issues with walking, frequent falls
- hyperCKemia, meaning elevated blood levels of the enzyme creatine kinase, a marker of muscle damage
- hyporeflexia (decreased or absent reflexes)
- tongue weakness, trouble chewing and swallowing, weight loss
- delayed motor development in children
- breathing problems, weak cough, and frequent respiratory infections
- sleep impairments such as daytime sleepiness and sleep apnea (when breathing stops and starts repeatedly during sleep).
Pompe Disease News is strictly a news and information website about the disease. It does not provide medical advice, diagnosis, or treatment. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website.
FAQs about Pompe disease symptoms
Across the different types of Pompe disease, nearly all patients experience muscle weakness. Breathing problems and difficulty eating also are common in people with a Pompe disease diagnosis. Almost all babies with the classic infantile-onset form of the disease develop an enlarged heart and heart disease.
Pompe disease causes are linked to genetic mutations that render the body unable to break down glycogen, a complex sugar molecule used by some cells as a form of short-term energy storage. Muscle cells, which require a lot of energy to power movement, normally store significant amounts of glycogen, thus muscles are primarily affected in Pompe. Cells of the heart and skeletal muscles (muscles responsible for the movement of body parts) are the most affected.
When a person breathes, muscles in the diaphragm work to push air into and out of the lungs. Pompe disease can cause weakness in these muscles, and likely also in other accessory respiratory muscles, which can lead to impaired breathing.
The speed of Pompe progression varies widely from person to person, depending on the type of disease as well as treatments and other factors. In general, an earlier onset of symptoms is associated with a faster disease progression.
Every person inherits two copies of the GAA gene, one from each biological parent. Pompe will only develop if both copies of this gene contain a disease-causing mutation. Carriers, who have only one mutated copy (and one healthy copy), will not develop Pompe symptoms, but they may pass the disease-causing mutation to their biological children.
Related Articles
-
Discussion
-
 Discussion
Discussion
-
Discussion
-
 Discussion
Discussion
-
Discussion
-
 Discussion
Discussion