
Treatment options for Pompe disease
 Fact-checked by
Fact-checked by Pompe disease treatment typically requires a multidisciplinary healthcare team, including neurologists, cardiologists, and respiratory therapists, among others. Currently, enzyme replacement therapy is the standard treatment for Pompe disease.
Pompe disease enzyme replacement therapy, or ERT, targets the underlying causes of the condition to slow its progression and potentially prolong survival. In addition, supportive treatments like physical, occupational, and speech therapy can help manage specific Pompe disease symptoms, improving quality of life for some patients.
Enzyme replacement therapies
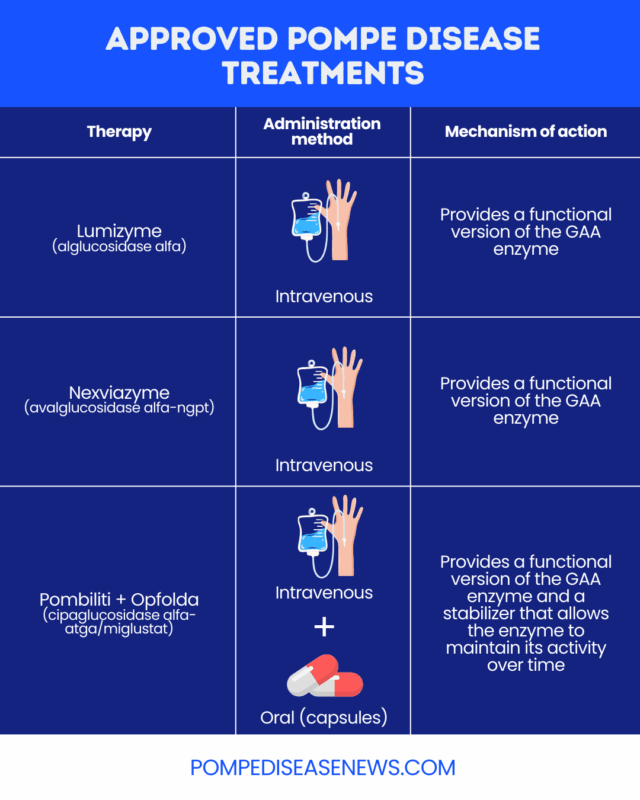
To date, ERT medications are the only approved Pompe disease treatment options.
ERT targets the underlying cause of Pompe disease: the lack or dysfunction of an enzyme called acid alpha-glucosidase (GAA). This happens as a result of mutations in the GAA gene, which provides instructions for cells to make the enzyme. Pompe disease is an autosomal recessive disorder, meaning that a person must inherit two mutated GAA copies, one from each biological parent, to develop the disease.
GAA normally helps break down a complex sugar called glycogen. Without functional GAA, glycogen builds up to toxic levels within cells, ultimately leading to symptoms such as progressive muscle weakness and difficulty breathing. ERT provides a functional version of the GAA enzyme, which can replace the missing or faulty enzyme, thereby slowing or preventing glycogen buildup in the body.
Approved ERT medications
Two ERT medications, both administered via into-the-vein (intravenous) infusions, have been approved by the U.S. Food and Drug Administration (FDA) for people with Pompe disease:
- Lumizyme (alglucosidase alfa): approved for children and adults with all types of Pompe disease
- Nexviazyme (avalglucosidase alfa-ngpt): approved as a late-onset Pompe disease treatment for patients 1 year and older
Another approved therapy for Pompe disease is the two-part combination therapy, Pombiliti + Opfolda (cipaglucosidase alfa-atga/miglustat). It includes a version of the GAA enzyme called cipaglucosidase alfa-atga (Pombiliti), which is administered intravenously, and an enzyme stabilizer, or chaperone, called miglustat (Opfolda) that’s taken orally. The stabilizer allows the enzyme to maintain its activity over time. Pombiliti + Opfolda is specifically approved for adults with late-onset Pompe disease who weigh at least 40 kg (about 88 pounds) and aren’t improving on their current ERT.
ERT in CRIM-negative patients
Some people with Pompe can’t make any GAA and are referred to as being cross-reactive immunologic material (CRIM)-negative, while others can produce it to some extent — although it doesn’t function properly — and are CRIM-positive. Generally, ERT is less effective for CRIM-negative patients, because their bodies can mistake the therapeutic enzyme for an infectious invader. The resulting immune response can interfere with the therapy’s function. To increase treatment efficacy, CRIM-negative patients may be given additional immune-modulating therapies alongside ERT.
Supportive therapies
Besides ERT, several long-term supportive interventions can help people with Pompe disease maximize their ability to function in day-to-day life. These may include:
- physical therapy to improve muscle strength, preserve mobility, and reduce muscle contractures
- speech therapy to strengthen facial muscles and facilitate speech and swallowing
- respiratory therapy to strengthen the muscles that push air in and out of the lungs and improve breathing
- occupational therapy to help develop strategies that make it easier to live with Pompe disease and find new ways to engage in daily activities
- dietary support with a high-protein, low-carbohydrate diet to ensure patients meet their nutritional needs, as well as supplements to support bone health
- mobility devices and orthopedic devices such as leg braces to make it easier to move around and support muscle function
The healthcare professionals who have expertise in these supportive interventions can also be helpful resources in other ways. For example, physical therapists can help patients develop strategies to avoid falls and use adaptive equipment, while speech therapists can assist patients in finding alternative methods of communication. Respiratory therapists can instruct patients on how to clear their airways, while dietitians can provide guidance about using feeding tubes for infants and older individuals with difficulty swallowing.
Experimental therapies
Several experimental therapies for Pompe disease are in various stages of development. Some focus on alleviating symptoms, while others aim to address the disease’s underlying causes. Therapeutic strategies under investigation for Pompe disease include:
- gene therapy: delivers a healthy copy of the GAA gene to body cells, allowing them to produce functional GAA enzyme
- substrate reduction therapy: blocks the production of glycogen to slow the toxic buildup that leads to symptoms of Pompe disease
- chaperone therapy: binds to the dysfunctional GAA enzyme, helping it fold properly and potentially restoring its activity
- lysosomal exocytosis activators: stimulate the process that allows lysosomes, which are like recycling centers in cells, to release their cargo (including glycogen) outside cells, thereby potentially preventing the toxic buildup of glycogen
It isn’t yet known how many people with Pompe disease may benefit from these experimental strategies. Some therapeutic approaches may work better for early- or late-onset types of the disease.
Chaperone therapy would also have limited applicability. About 10% to 15% of Pompe disease patients have mutations that result in the production of versions of GAA that a chaperone can act on, though it is unclear how this will translate to clinical efficacy.
The FDA-approved combination therapy Pombiliti + Opfolda contains a chaperone (Opfolda), but no chaperone therapies are currently approved as a standalone treatment for Pompe disease.
How to treat Pompe disease: Step-by-step care pathway
The details of a Pompe disease care plan will vary between individuals, depending on disease type and other factors. Patients should always discuss the specifics of their treatment with their doctors to better understand the steps and timeline of their treatment pathway.
A typical pathway can include:
- Confirming a Pompe disease diagnosis and identifying its subtype
- Assessing muscle, heart, and respiratory function
- Determining CRIM status to assess the risk of an immune response that may neutralize ERT. In CRIM-negative patients, additional immune-modulating therapies may be needed to increase ERT efficacy.
- Starting ERT, generally as early as possible, although the exact timing depends on disease type and symptoms
- Introducing supportive therapies
- Regularly reassessing symptoms and function, and adjusting treatment according to medical guidance as needed
Is there a cure for Pompe disease?
There is no cure for Pompe disease. However, ERT can slow disease progression and improve patients’ functional ability.
Pompe disease life expectancy with treatment
Pompe disease life expectancy with treatment is longer than would be expected without treatment. However, specific survival outcomes vary widely, depending on the type and severity of the disease and the timing of ERT initiation. Typically, starting ERT earlier leads to better survival outcomes.
Without treatment, babies with classic infantile-onset Pompe disease typically die in the first year or two of life. However, studies have shown that Lumizyme can extend survival and reduce the risk of death by about 90%. Although more patients are living longer with treatment, long-term support needs may still be high.
Lifespan in untreated late-onset Pompe disease can range widely depending on when symptoms appear, how quickly they progress, the rate of respiratory failure, and other coexisting disorders. A 2016 review study with data from 438 patients found that treatment with Lumizyme reduced mortality by nearly 5 times compared with patients who didn’t receive treatment.
Does the type of Pompe disease affect treatment?
ERT treatment regimens differ for people with the classic infantile-onset form of Pompe disease and those with later-onset forms. In some countries, individual ERT medications may be approved only for certain types of Pompe disease, not others.
Experts typically recommend that classic infantile-onset Pompe disease treatment begin as soon as possible after diagnosis. People with late-onset Pompe disease usually begin ERT once they start showing detectable signs of muscle weakness, with follow-up assessments every six months.
Pompe Disease News is strictly a news and information website about the disease. It does not provide medical advice, diagnosis, or treatment. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website.


