
 Discussion
Discussion
Pompe disease overview
Last updated Nov. 19, 2024, by Marisa Wexler, MS
 Fact-checked by José Lopes, PhD
Fact-checked by José Lopes, PhD
Pompe disease is a rare genetic condition that is characterized by the abnormal buildup, inside cells, of a complex sugar molecule called glycogen. This buildup impairs the workings of different organs and tissues, especially the heart and other kinds of muscle.
Pompe disease is also known as glycogen storage disease type 2, or GSD2, and acid maltase deficiency.
What are the symptoms of Pompe disease?
Pompe disease symptoms among patients can vary greatly, based on the person’s age at disease onset, the type of Pompe disease, and the rate of disease progression and severity.
The most common symptoms associated with Pompe disease are:
- progressive muscle weakness
- poor muscle tone
- breathing problems
- respiratory infections
- trouble eating
- enlargement of the tongue, the liver, called hepatomegaly, and/or heart, known as cardiomegaly
- hearing impairment.
Read more about Pompe symptoms
What are the types of Pompe disease?
Pompe disease is classified into three types:
- classic infantile-onset form
- nonclassic infantile-onset form
- late-onset form.
Classic infantile-onset Pompe disease
Babies with classic infantile-onset Pompe disease typically start to exhibit symptoms — including difficulty feeding, poor muscle tone, and delayed motor development — within the first few months after birth. Heart muscle disease, known as cardiomyopathy, usually develops within the first weeks of life. Difficulty breathing and hearing loss also are common.
Nonclassic infantile-onset Pompe disease
In this form of Pompe disease, symptoms become apparent within the first year of life, with babies showing muscle weakness and delayed motor development. Symptoms of Pompe disease in infants with the nonclassic infantile-onset form usually develop later than in the classic form. Also, unlike the classic form, babies with nonclassic infantile-onset Pompe do not usually develop cardiomyopathy within the first year of life. For this reason, some classification systems consider this form of Pompe disease to be a subtype of the late-onset form.
Late-onset Pompe disease
Late-onset disease refers to Pompe when symptoms develop at any point after the first year of life. Some people with late-onset Pompe disease develop symptoms in childhood, while others only experience symptoms of Pompe disease as adults or in the latter decades of life — which is the reason why this type is sometimes called adult-onset Pompe disease.
Generally, a younger age at onset is associated with more severe disease. People with late-onset Pompe disease typically experience progressive muscle weakness, especially affecting the torso and legs. Breathing problems also may develop as the disease progresses.
What are the causes of Pompe disease?
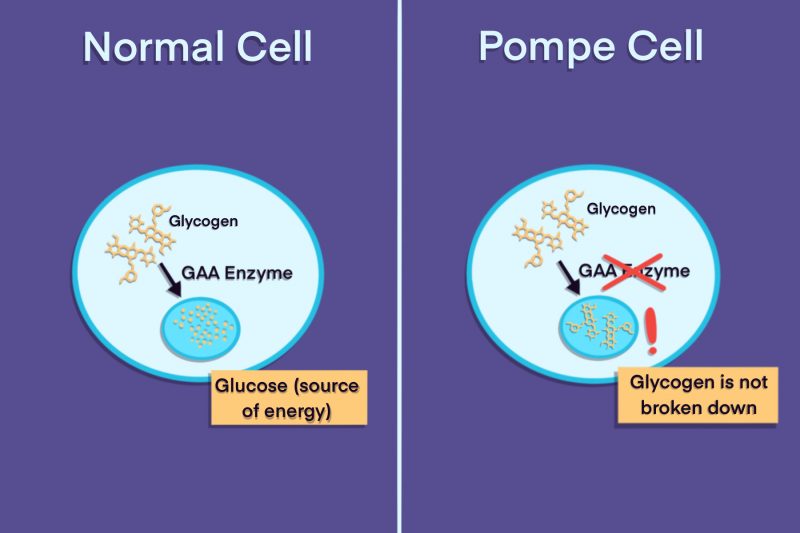
Pompe disease is caused by mutations in the GAA gene, which encodes instructions for making the acid alpha-glucosidase (GAA) enzyme. A mutation in the GAA gene can either lead to the production of a GAA enzyme that doesn’t work correctly, or prevent the production of the enzyme entirely.
This enzyme breaks down a large molecule called glycogen into glucose when the body needs energy. Glycogen is a form of sugar the body stores mainly in cells of the liver and skeletal muscles, where it works as a reserve of energy.
In people with Pompe disease, glycogen cannot be broken down and builds to toxic levels inside cells. Muscle cells are especially affected because glycogen normally serves as a main source of energy to power muscle movements.
How is Pompe disease treated?
To ensure all aspects of the condition are treated, Pompe disease patients usually are cared for by a multidisciplinary team of specialists. This team typically comprises cardiologists, neurologists, pulmonologists, respiratory therapists, dietitians, orthopedists, occupational/speech therapists, geneticists, and genetic counselors.
Disease-specific medications are a standard approach to Pompe disease treatment. In that regard, enzyme replacement therapy, or ERT, provides Pompe patients with an engineered version of the GAA enzyme to replace the missing or nonworking enzyme.
Two ERTs for Pompe disease are widely available; both are sold by Sanofi. They are Lumizyme (alglucosidase alfa), sold under the brand name Myozyme outside the U.S., and Nexviazyme (avalglucosidase alfa). Lumizyme is approved for all types of Pompe disease, while Nexviazyme is a next-generation therapy approved for patients ages 1 and older in the U.S. The approved age ranges in other locations vary.
In addition, Amicus Therapeutics’ combination therapy Pombiliti (cipaglucosidase alfa) + Opfolda (miglustat) has been approved in the U.S. and other markets for certain adults with late-onset Pompe disease. Cipaglucosidase alfa (Pombiliti) is a form of the GAA enzyme, while miglustat (Opfolda) is an enzyme stabilizer.
Healthcare providers also can prescribe supportive therapy to help manage specific Pompe symptoms. This may include physical therapy to strengthen muscles and a person’s overall physical condition, as well as occupational therapy, and speech therapy.
Read more about Pompe treatment
How is Pompe disease inherited?
All people inherit two copies of the GAA gene — one from each biological parent. Pompe disease inheritance occurs in an autosomal recessive fashion, meaning that the disease will only develop in offspring if both copies of the GAA gene are mutated. A person with only one mutated copy is referred to as a Pompe disease carrier. These individuals will not develop the disease, but may pass the Pompe-causing mutation to their biological children.
If two carriers have biological children, there is a 1 in 4 (or 25%) chance that each child would inherit both mutated copies of the gene and develop Pompe disease. Likewise, there is a 25% chance that any child would inherit two normal GAA genes, and so would not have the disease nor be a carrier. There is a 50% chance that any children would inherit one mutated gene and one normal gene — so the children would be carriers like their parents.
Read more about Pompe inheritance
How is Pompe disease diagnosed?
A diagnosis of Pompe disease can first be suggested by clinical findings, particularly those related to breathing problems and evidence of muscular weakness. A differential diagnosis — one that distinguishes between different diseases with similar symptoms — is important, because Pompe disease may be wrongly assumed to be another disorder that causes muscle wasting, and thus be misdiagnosed.
Confirming a Pompe disease diagnosis usually involves enzymatic tests to measure the activity of the GAA enzyme. In children with infantile-onset Pompe disease, the activity of the GAA enzyme is typically less than 1% of normal. In individuals with the late-onset form of this disease, GAA activity is usually 40% of normal or lower.
Genetic testing to look for disease-causing mutations in the GAA gene can help confirm the diagnosis, and is particularly helpful in identifying carriers. For people who are known carriers of the disease, genetic testing before the baby is born, also known as prenatal testing, may be available and help ensure an early diagnosis.
Read more about Pompe diagnosis
What is the prognosis of Pompe disease?
Pompe disease life expectancy, and a prognosis, can vary substantially from person to person, depending on the individual’s disease type and the specific clinical characteristics. Generally, mutations that lead to greater functional impairment of the GAA enzyme are associated with more severe disease. Specifically, when Pompe disease causesworse impairments, it is generally tied to an earlier age at onset and faster symptom progression.
Without treatment, babies with classic infantile-onset Pompe disease typically don’t survive past age 2, and most children with the nonclassic infantile-onset form survive only into early childhood. In the classic type, symptoms usually begin within a few months of birth. The prognosis of Pompe disease in adults, and in children with the late-onset type, is largely dependent on the extent to which the disorder causes breathing problems.
Novel treatments such as ERT or Pombiliti + Opfolda are improving outcomes for people with all forms of Pompe disease. However, specific Pompe treatments have only been available for a decade or two, so the long-term prognostic outlook for treated patients is still not completely understood.
Read more about Pompe life expectancy
What other conditions could Pompe disease be mistaken for?
A number of other health conditions may cause muscle weakness and other symptoms commonly associated with Pompe. These include other glycogen storage disorders, which, like Pompe disease, are characterized by a toxic buildup of glycogen. Other disorders affecting muscle health may also be associated with Pompe disease.
In babies with infantile-onset forms of Pompe disease, symptoms may resemble:
- glycogen storage disease type 3a, also called debrancher deficiency
- glycogen storage disease type 4, also known as branching enzyme deficiency
- heart diseases such as myocarditis, characterized by inflammation in the heart’s muscle
- spinal muscular atrophy type 1
- Danon disease
- carnitine uptake disorder
- disorders that impair the activity of mitochondria, the powerhouse of the cell.
Pompe disease symptoms in adults and in children with late-onset Pompe disease may resemble:
- glycogen storage disease type 5, known as McArdle disease
- glycogen storage disease type 6, also called Hers disease
- muscular dystrophies, a group of genetic disorders that cause progressive muscle weakness
- polymyositis, a chronic inflammatory disease.
Pompe Disease News is strictly a news and information website about the disease. It does not provide medical advice, diagnosis, or treatment. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website.
FAQs about Pompe disease
Despite the approvals of specific therapies for Pompe disease, no cure is available. Still, Pompe patients can benefit from enzyme replacement therapies, designed to slow the progression of the disease by reducing the toxic buildup of glycogen inside cells, as well as from the combination therapy Pombiliti + Opfolda. Supportive treatments like physical therapy and occupational therapy also can be of help in navigating life with Pompe disease.
The estimated incidence (the number of new cases in a given time period) of Pompe disease varies in different locations and among ethnic groups. In the U.S., Pompe disease affects about 1 of every 40,000 people overall, though the rate is higher — 1 of every 14,000 — among African Americans. Among people of European descent, late-onset Pompe affects 1 in every 60,000 people, while infantile-onset Pompe affects about 1 in 100,000 individuals. In southern China and Taiwan, the incidence of Pompe disease has been estimated at 1 in every 50,000 people, while in Australia, the estimated incidence is 1 in 145,000.
In the U.S., the federal government recommends checking newborns for a number of serious genetic disorders that can be tested for shortly after birth; these diseases are listed on the recommended uniform screening panel (RUSP), to which Pompe was added in 2015. While the U.S. government recommends screening for all disorders on the RUSP, each state ultimately decides what conditions are tested for in that state’s newborn screening programs. As of 2023, Pompe disease was a part of newborn screening programs in 45 U.S. states and Washington, D.C.
Pompe disease is a lifelong and progressive disorder, and its symptoms generally worsen over time. Disease symptoms often can cause difficulties with activities in day-to-day life — for example, most late-onset patients experience some difficulty walking, and many will eventually rely on a wheelchair to get around. Learning new coping strategies and finding accommodations can help make navigating daily life with Pompe disease less burdensome.
All forms of Pompe disease are life-limiting — the disease can cause problems with the heart muscle and muscles needed for breathing, leading to heart and respiratory failure. The specific prognostic outlook varies depending on a number of factors, including the disease type, specific disease-causing mutations, and treatments.
Related Articles
-
Discussion
-
 Discussion
Discussion
-
 Discussion
Discussion
-
Discussion
-
 Discussion
Discussion
-
Discussion